

22 Nov Biosimilars hurdles in safety monitoring
Biological products (BP), or biologics, is a class of heterogeneous medical products derived from different natural source materials (mammalian, insect and plant cell cultures, yeast, etc.) intended to treat, cure, prevent or diagnose diseases and medical conditions [1, 5]. BP introduction to pharmaceutical market has revolutionized treatment strategy for many therapeutic areas such as oncology [2], dermatology, rheumatology, etc. helping to fight with rheumatoid arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, psoriasis [5]. In spite of diversity these products may be divided into different groups according to, for instance, following features – origin (plant, mammalian, bacterium, etc.), indication (antirheumatic, antipsoriatic, etc.), and molecular size. If classified by latter parameter one of the most notable and not only by high molecular weight and structure complexity are monoclonal antibodies (mAb). Although mAb present comparatively new drug class patent protection for some of them has already expired (e.g. adalimumab, abciximab, bevacizumab, etanercept, infliximab, omalizumab, ranibizumab, rituximab) [4] or expires in the nearest future causing plenty of opportunities for production of follow-on biologics (more commonly called “biosimilars”) [3].
Biosimilars are “copies” of original drugs that are just similar, not identical to them. Such difference may be explained by inability to exactly copy manufacturing process of original biologics, which in terms results from lack of available information published by developer of the original product, necessity for reversed engineering, absence of reliable analytical methods allowing original product and biosimilar comparison, molecule complexity and some other factors. Unfortunately, it seems rather complicated to increase likeness of follow-on and original biologics and change status quo not only due to described reasons, but because of variability in regulatory requirements as well.
General guidelines for the approval of biosimilars have been developed or are being drafted by a large number of countries. However, up to this moment in clinical and post-marketing requirements are diverse and sometimes rather vague for follow-on biologics in different countries (table 1).
Table 1
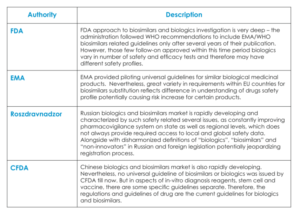
However, during second decade of 21st century EMA has already published separate “Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues” and approved biosimilars for monoclonal antibody drugs. These facts emphasize high importance of making quick decisions regarding these drugs making obvious requirement for following global upgrades:
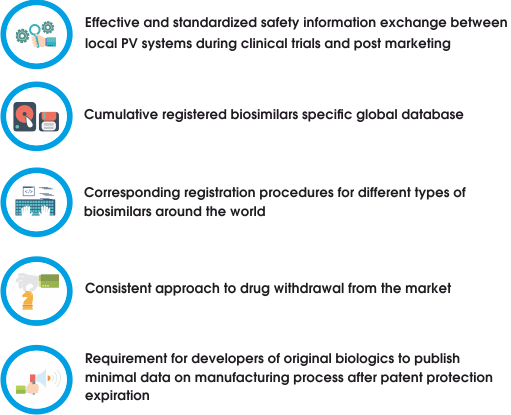
Considering global trends for discussions and enhanced information exchange within global biologics society there is a hope that European experience will be improved and (like EMA/WHO guidelines for biosimilars) translated to all other countries resulting in increase of mAb and other biosimilars likeness, safety and efficacy.
The full text download here article-about-biosimilars
No Comments